Множественный дефицит гормонов гипофиза (или пангипопитуитаризм) характеризуется недостатком соматотропного гормона (СТГ), дефицитом тиреотропного (ТТГ) и адренокортикотропного гормона (АКТГ), а также лютеонизирующего (ЛГ) и фолликулостимулирующего (ФСГ) гормонов и в некоторых случаях низким уровнем пролактина.
Пангипопитуитаризм может быть врожденным (дефект генов) или приобретенным.
Наследственный множественный дефицит гормонов гипофиза обусловлен мутациями в генах POU1F1 (PIT-1), PROP-1, LHX-3, LHX-4, HESX-1, Pitx2.
Основные причины приобретенного множественного дефицита гормонов гипофиза:
1. Опухоли гипоталамуса и гипофиза:
- краниофарингиома;
- гамартома;
- нейрофиброма;
- герминома;
- аденома гипофиза.
2. Опухоли других отделов мозга
3. Травмы:
- черепно-мозговая травма;
- хирургическое повреждение гипофизарной ножки.
4. Инфекции:
- энцефалит и менингит;
- неспецифический (аутоиммунный) гипофизит.
5. Супраселлярные арахноидальные кисты, гидроцефалия, симптом «пустого турецкого седла».
6. Патология сосудов головного мозга (кровоизлияния)
7. Облучение головы и шеи при онкологических заболеваниях
8. Токсические последствия химиотерапии.
9. Инфильтративные болезни:
- гистиоцитоз;
- саркоидоз.
10. Транзиторный:
- конституциональная задержка роста и пубертата;
- психосоциальный (депривационный) нанизм.
Важно!
Для детей с дефицитом СТГ на фоне объемного образования в головном мозге (краниофарингиомы) или после перенесенной травмы головы характерны более поздние сроки проявления дефицита роста — после 5–6-летнего возраста. У детей с объемным образованием головного мозга резко снижается скорость роста, может появиться полидипсия (много пьет) и полиурия (выделяет много мочи), при этом выделяет неконцентрированную мочу. Полиурия и полидипсия являются характерными признаками развития центрального несахарного диабета в результате развития синдрома сдавления гипофизарной ножки и нарушения выделения антидиуретического гормона.
Наличие помимо дефицита роста таких жалоб, как головные боли, нарушение зрения, рвота, позволяет заподозрить внутричерепную патологию (краниофарингиома).
У детей с транзиторным СТГ-дефицитом (конституциональная задержка роста и пубертата) семейный анамнез позволяет в большинстве случаев выявить аналогичные случаи низкорослости и задержки полового развития в детском и подростковом возрасте у одного из родителей либо ближайших родственников.
Для детей с врожденным множественным дефицитом гормонов гипофиза характерно замедление динамики роста с первых лет жизни. У детей с дефицитом СТГ скорость роста не превышает 4 см в год, чаще всего она составляет 1–2 см в год.
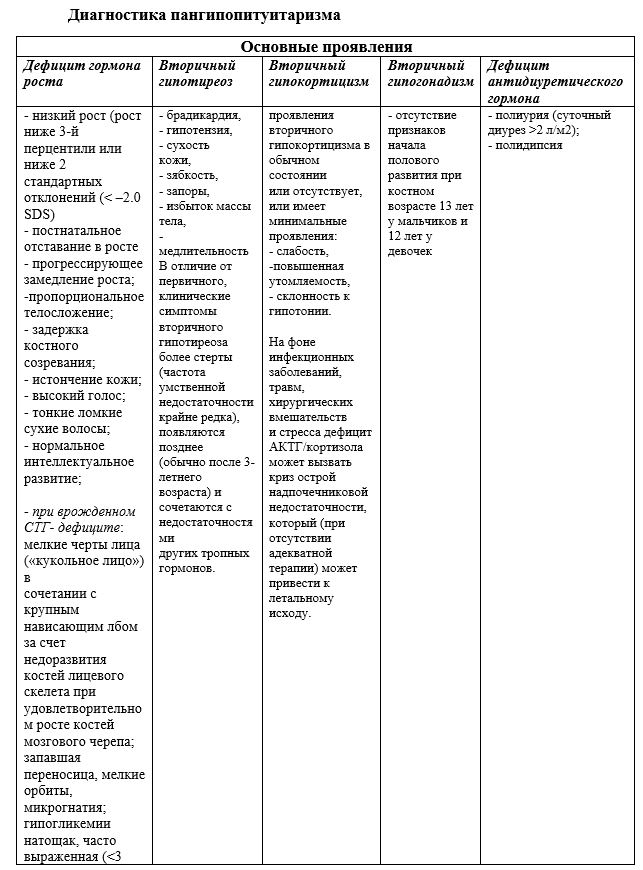
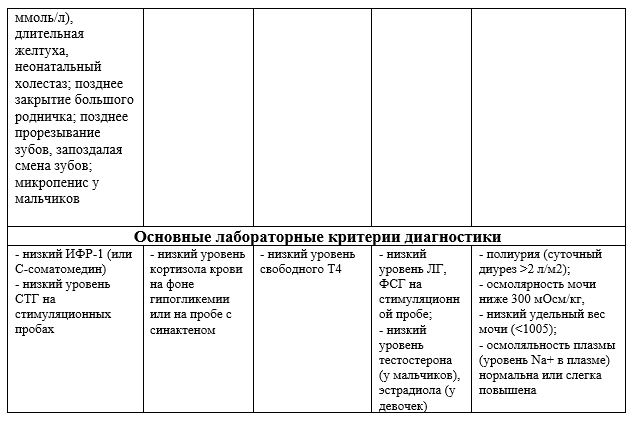
Кроме исследования показателей гормонального профиля, оценки костного возраста ребенку проводится МРТ головного мозга. В случаях подозрения на врожденный характер заболевания показано проведение молекулярно-генетического исследования с целью поиска мутаций в генах.
После постановки диагноза пангипопитуитаризм назначается лечение рекомбинантным гормоном роста, препаратами левотироксина, глюкокортикоидами. При выявлении несахарного диабета проводится лечение синтетическим аналогом антидиуретического гормона.
Дефицит пролактина характерен для пациентов с мутациями в гене PIT-1 и PROP-1. У детей и подростков недостаточность пролактина не имеет клинических проявлений, лечение не проводится.
При диагностике объемного образования головного мозга решается вопрос о проведении хирургического лечения.